TLA+ conference 2024

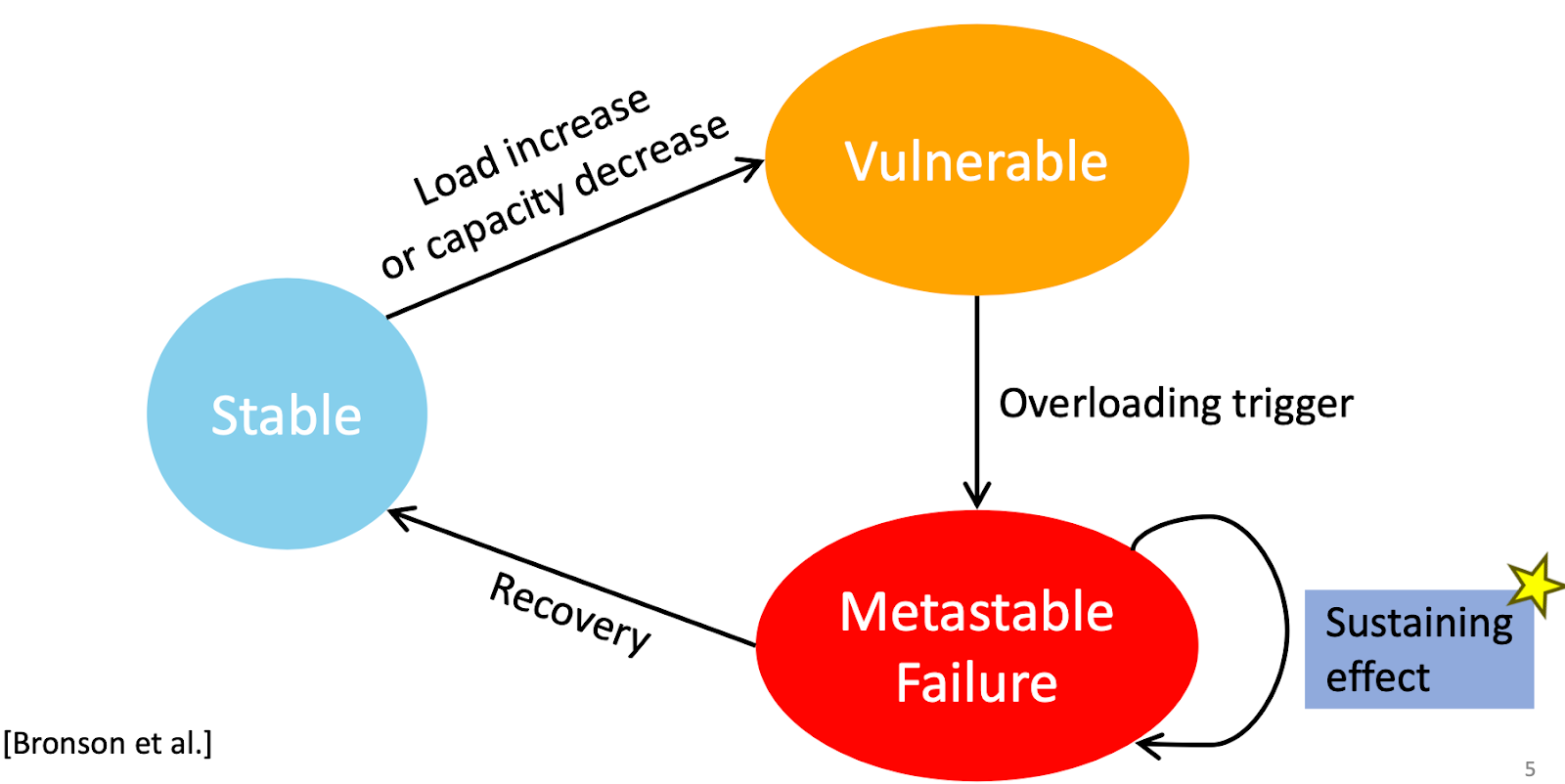
The TLA+ foundation got established last year in April as an independent, non-profit organization under the umbrella of the Linux Foundation. This April 15th, we colocated our annual TLA+ conference with the Linux OSSNA event to reach out to developers attending this event. Being under Linux Foundation, we didn't have to pay for the venue, but we paid $3K for professional recording of the sessions, so we can share the talks with the world at large. The videos will be available within a couple weeks. Marc Brooker: 15 years of TLA+ Marc Brooker gave the keynote this Monday in TLA+ conference 2024. His keynote was a good mix of inspiration for formal methods and challenging formal methods to do better. Brooker is a VP/distinguished engineer at AWS. He has been on-call for 15 years, and were involved with many popular AWS services, including Aurora, Lambda, EC2, EBS, API gateway, iot, bedrock. He defines himself as mostly a practitioner, building large scale distributed systems. Br